














[캠퍼스人+스토리]
새로운 응용 분야 개척 등 발전
새로운 응용 분야 개척 등 발전
|
|

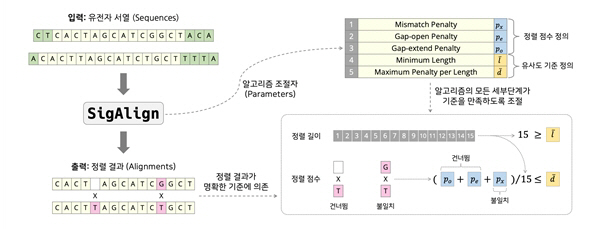
유전체 분석은 과정뿐 아니라, 대규모의 생물정보 데이터 처리 단계가 필수적이다. 한 사람당 30억개의 염기서열을 분석하는 유전체 자료는PB (페타바이트) 규모에 달한다. 염기쌍을 효율적이고 빠르게 찾는 알고리즘은 매우 중요한 원천기술이다.
서울대 연구진이 개발한 'SigAlign'은 널리 사용되고 있는 알고리즘 (BWA-MEM, HISAT2, Bowtie2) 등에 비해 수십 배 빠른 속도로 더 정확한 결과를 도출했다. 표준 컴퓨팅 환경에서의 방대한 비교분석에서 초당 10만 개 이상의 데이터를 처리할 수 있는 유일한 알고리즘이다.
SigAlign은 기술적으로도 매우 효율적인 메모리 사용이 가능하도록 개발되어, 고성능 컴퓨터 (CPU 100개, 메모리 1-2 TB)가 필요했던 기존의 알고리즘과는 달리 향후 휴대폰이나 소형 IoT기기에서도 사용이 가능한 분석 알고리즘이다.
연구진은 "많은 사람들이 연구성과를 활용해 더 좋은 연구를 효율적으로 수행하게 하기 위한 선택"이라며 "알고리즘이 공개된다고 해도 이를 활용한 다양한 솔루션의 개발은 난이도가 높은 작업으로 연구진이 지속적으로 주도권을 가지고 새로운 응용 분야를 개척하고 발전시킬 것"이라고 강조했다.

